主成份阐明(PCA)在生物芯片样本筛选中的应用及在R语言中的实现
主成份阐明要领可以对基因芯片的样本聚类环境举办可视化,可得到样本在尝试组和比较组之间的直观漫衍环境,从而便于对异常样本举办检测和去除,不然异常样本的存在将会对差别基因的判断等后续阐明造成倒霉影响。下面我将演示在R语言中如何对芯片样本举办PCA阐明及可视化。
首先,需要下载R语言的措施,R是一个很好的免费统计语言,官方下载网址为:www.r-project.org/
为了便于演示,我首先用在R中成立一个模仿的芯片数据矩阵,该矩阵为10000行(10000个基因),30列(30个样本):
chip.dat<-matrix(rnorm(10000*30,mean=0),ncol=30,nrow=10000)
我把30个样天职为两组,前15列和后15列各为一组,给它们界说差异的颜色:
colour<-c(rep(2,15),rep(3,15))
在10000个基因中,我们假定有100个基因在两个组间是有差此外,我们假设个中有50个在前一组是上调的,另50个在前一组中是下调的:
diff.ind<-sample(1:10000,100)
chip.dat[diff.ind[1:50],1:15]<-rnorm(50*15,mean=2)
chip.dat[diff.ind[51:100],1:15]<-rnorm(50*15,mean=-2)
如此,我们就结构好了这个模仿数据,虽然,这个数据中两组间的样本是没有问题的,也就是说没有异常值的,下面我们可以对其举办PCA阐明:
chip.dat.pca<-princomp(chip.dat)
然后我们可以对前三个主成份举办可视化,看看这30个样本在前三个主成份的空间中的漫衍,这需要用到rgl包中的plot3d的函数:
library(rgl)
plot3d(chip.dat.pca$loadings[,1:3],col=colour,type=”s”,radius=0.025)
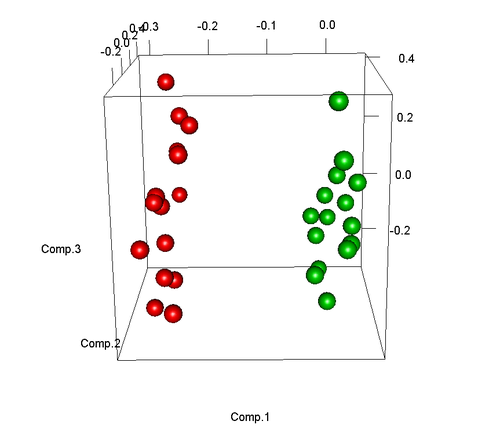
这样我们就可以获得了三维的样天职布图如下:
可是,假如样本中有异常样本,那么PCA也可以很好的发明,下面将会模仿样本15为异常样本(如尝试未乐成而其实因该属于比较组):
可是,假如样本中有异常样本,那么PCA也可以很好的发明,下面将会模仿样本15为异常样本(如尝试未乐成而其实因该属于比较组):
chip.dat<-matrix(rnorm(10000*30,mean=0),ncol=30,nrow=10000)
chip.dat[diff.ind[1:50],1:14]<-rnorm(50*14,mean=2)
chip.dat[diff.ind[51:100],1:14]<-rnorm(50*14,mean=-2)
chip.dat[diff.ind[1:50],1:14]<-rnorm(50*14,mean=2)
chip.dat[diff.ind[51:100],1:14]<-rnorm(50*14,mean=-2)
对该数据举办PCA阐明及可视化:
chip.dat.pca<-princomp(chip.dat)
plot3d(chip.dat.pca$loadings[,1:3],col=colour,type=”s”,radius=0.025)
chip.dat.pca<-princomp(chip.dat)
plot3d(chip.dat.pca$loadings[,1:3],col=colour,type=”s”,radius=0.025)
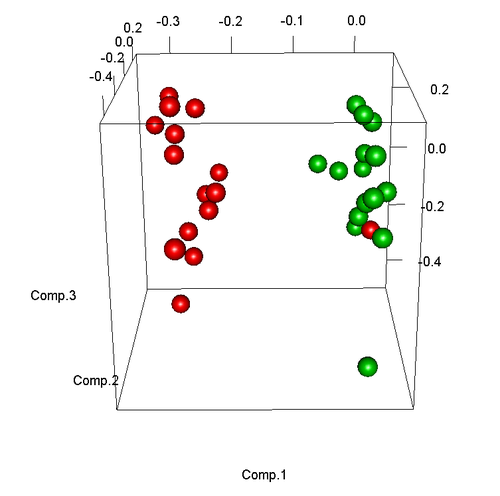
呈现下图:

关键字:
